

When a newborn arrives, parents expect those first magical moments to be filled with joy and wonder. But for some families, these moments are overshadowed by serious health concerns seemingly out of nowhere. Acute liver failure in infant cases represents one of the most challenging medical emergencies that can affect our littlest ones. Unlike adults, whose liver issues often develop after years of various exposures or lifestyle factors, babies can experience rapid liver deterioration due to genetic blueprints they’ve inherited before they’ve even taken their first breath.
The liver, that remarkable chemical factory performing over 500 essential functions, shouldn’t typically show signs of struggle in those earliest days of life. Yet for approximately 1 in 2,500 newborns, genetic conditions affecting the liver become apparent, sometimes with frightening speed. These acute liver failure in infant causes can be bewildering for parents and challenging even for experienced medical professionals to diagnose quickly. Understanding these hereditary disorders is crucial not just for affected families but for anyone who might someday face the worry of unexplained infant jaundice that doesn’t fade, unusual bleeding, or a baby who simply isn’t thriving. Today, we’ll explore the main types of inherited conditions that can lead to this serious situation, explaining them straightforwardly while offering insight into how medical science is working to give these tiny fighters a fighting chance.
Understanding Infant Liver Failure: The Basics
Before diving into specific disorders, let’s understand what liver failure in babies means:
- The liver loses its ability to perform essential functions
- Can develop suddenly (acute) or gradually (chronic)
- Affects approximately 1 in 15,000-20,000 live births1
- Requires urgent medical attention and evaluation
- Has different causes and presentation from adult liver failure
Acute liver failure in newborn babies represents a medical emergency that requires immediate specialist attention. Unlike adult liver failure, which often develops after years of damage, infant liver failure can appear within days or weeks of birth when caused by hereditary factors.
When a baby’s liver begins to fail, it can no longer effectively remove toxins from the blood, process nutrients, produce proteins needed for growth and blood clotting, or store essential vitamins and minerals. The effects ripple throughout the tiny body quickly, as the liver touches virtually every metabolic process. What makes infant liver failure particularly challenging is that babies cannot communicate their discomfort besides crying, and early symptoms may be subtle or resemble other common infant issues.
Table 1: All you need to know about the basics of acute liver failure in infant.
| Category | Details |
|---|---|
| What is Infant Liver Failure? | A rare but serious condition where an infant’s liver loses its ability to function properly. It can happen suddenly or develop over time. |
| Functions of a Healthy Liver | – Filters toxins from the blood – Produces bile for digestion – Stores vitamins and minerals – Regulates blood clotting |
| Common Causes | – Genetic or metabolic disorders – Viral infections (e.g., hepatitis) – Autoimmune conditions – Reactions to certain medications or toxins |
| Early Symptoms | – Yellowing of the skin or eyes (jaundice) – Poor feeding – Irritability – Vomiting – Lethargy or sleepiness |
| Progressive Symptoms | – Swelling of the abdomen – Easy bruising or bleeding – Dark urine or pale stools – Failure to gain weight or grow |
| Diagnosis Methods | – Blood tests (to assess liver enzymes and function) – Ultrasound imaging – Liver biopsy (in some cases) – Genetic testing if needed |
| Possible Treatments | – Supportive care (nutrition, fluids, medications) – Treating the underlying cause – Liver transplant in severe cases |
| Importance of Early Detection | Prompt diagnosis and treatment can prevent complications and improve outcomes. Regular pediatric checkups can help catch early signs. |
| Long-term Outlook | Depends on the cause and severity. Some infants recover fully; others may need lifelong management or a liver transplant. |
| Parental Tips | – Watch for signs like jaundice and poor feeding – Keep all medical appointments – Communicate concerns quickly with your pediatrician |
For parents, the first signs often include prolonged jaundice (yellowing of the skin and eyes) that doesn’t improve by two weeks of age, unusual bleeding or bruising, swelling in the abdomen, or poor weight gain despite normal feeding. These baby liver problem symptoms should never be ignored, as early diagnosis dramatically improves outcomes. With advances in genetic testing, many hereditary causes can now be identified within days, allowing for targeted treatments that were impossible just a decade ago.
Also Read- Can You Get More Than One Autoimmune Disease?
Biliary Atresia
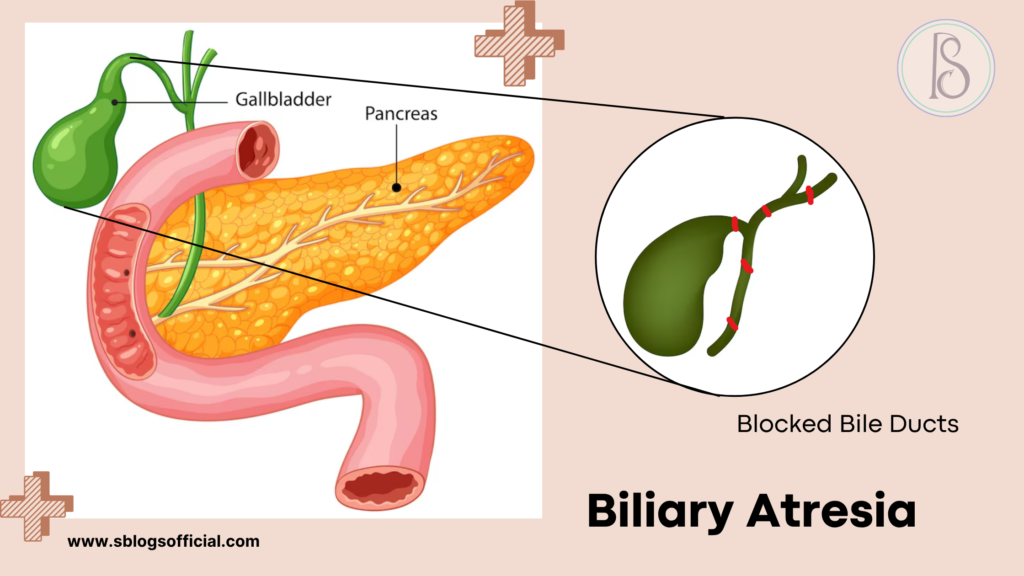
Biliary atresia represents one of the most serious liver conditions affecting newborns, fundamentally disrupting the normal flow of bile from the liver to the intestines. This devastating condition occurs when the bile ducts—tiny tubes responsible for carrying bile—are either completely absent or severely blocked, creating a cascade of liver complications. The condition typically manifests within the first few weeks of life, making early recognition crucial for infant survival. Unlike temporary jaundice that many newborns experience, biliary atresia causes progressive and irreversible liver damage if left untreated. Understanding this condition is essential for parents and healthcare providers, as prompt intervention can mean the difference between life and death for affected infants.
- What is it? A rare condition in infants where bile ducts are blocked or absent, preventing bile from flowing from the liver to the intestine.
- Key symptoms: Yellowing of the skin and eyes (jaundice), dark urine, pale or clay-colored stools, slow weight gain, enlarged liver.
- Why it matters: If untreated, it leads to progressive liver damage, cirrhosis, and eventual liver failure within the first two years of life.
- Clinical note: It affects approximately 1 in 10,000–15,000 live births and often requires surgical intervention (Kasai procedure) or liver transplantation.
Also Read- mRNA Vaccines: Future of Medicine – Let’s explore!
Tyrosinemia
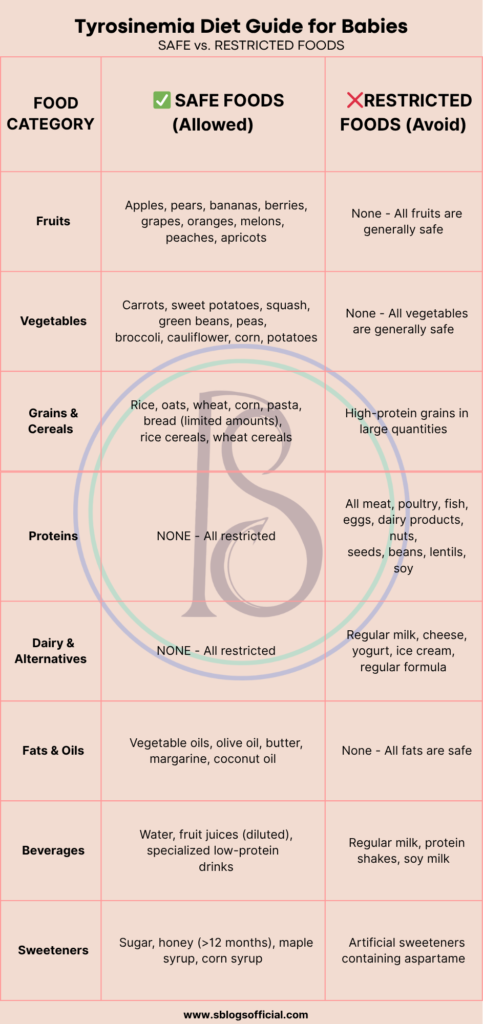
Tyrosinemia belongs to a group of inherited metabolic disorders that disrupt the body’s ability to process proteins properly, specifically affecting the breakdown of the amino acid tyrosine. When tyrosine cannot be metabolized correctly, it accumulates to toxic levels throughout the body, particularly targeting the liver, kidneys, and nervous system. This condition stems from genetic mutations that affect specific enzymes in the tyrosine degradation pathway, leading to a buildup of harmful byproducts. The disorder exists in three main types, with Type I being the most severe and life-threatening form. Early detection through newborn screening programs has revolutionized outcomes, as prompt treatment can prevent the devastating complications that once made this condition universally fatal.2
- What is it? A genetic disorder where the body cannot properly break down the amino acid tyrosine, leading to toxic buildup in the liver and other organs.
- Key symptoms: Failure to thrive, vomiting, diarrhea, cabbage-like odor, enlarged liver, bleeding tendencies, developmental delays.
- Why it matters: Without treatment, it can cause severe liver damage, kidney problems, and neurological complications that can be life-threatening.
- Clinical note: Type I is the most severe form, affecting 1 in 100,000 births, with higher rates in certain populations like French Canadians and Scandinavians.
Also read- Gluten Intolerance vs Lactose Intolerance | A Complete Guide
Alagille Syndrome
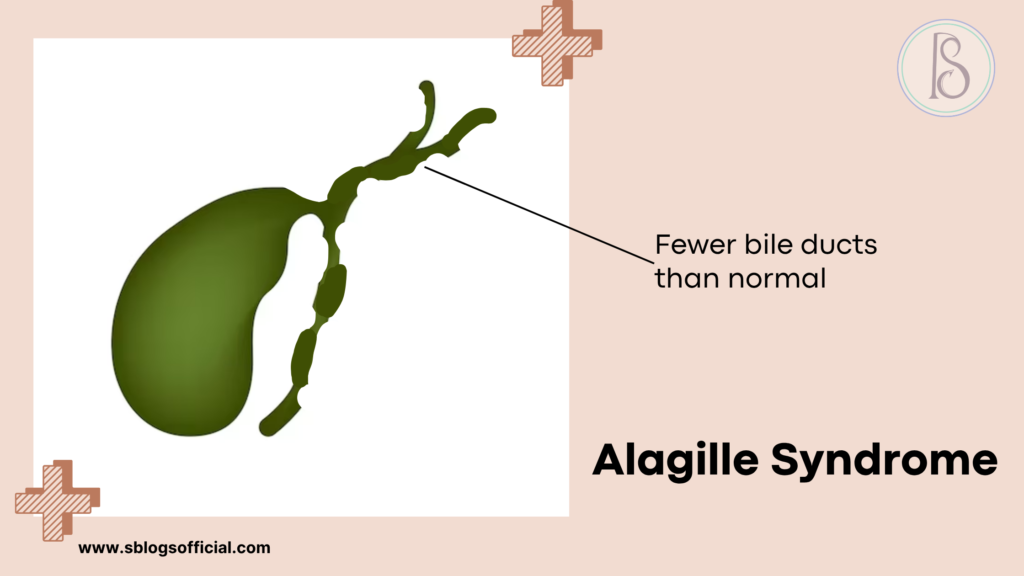
Alagille syndrome represents a complex multisystem genetic disorder that primarily affects liver development and function, though its impact extends far beyond hepatic complications. Named after French pediatrician Daniel Alagille who first described the condition in 1969, this syndrome results from mutations in genes crucial for normal organ development during embryonic growth. The hallmark feature involves a significant reduction in the number of bile ducts within the liver, leading to impaired bile flow and subsequent liver dysfunction. What makes Alagille syndrome particularly challenging is its variable expression—even within the same family, affected individuals can experience vastly different symptoms and severity levels. The condition affects multiple organ systems simultaneously, including the heart, skeleton, eyes, and kidneys, requiring comprehensive medical management throughout a patient’s lifetime.3
- What is it? A genetic disorder that affects the liver, heart, and other organs, characterized by fewer bile ducts than normal in the liver.
- Key symptoms: Chronic jaundice, itching, growth delays, heart murmurs, distinctive facial features, butterfly-shaped vertebrae.
- Why it matters: It can lead to progressive liver disease, heart problems, and developmental issues requiring lifelong management and monitoring.
- Clinical note: Affects approximately 1 in 30,000 live births, caused by mutations in JAG1 or NOTCH2 genes, with variable severity even within families.
Also read- Can I Gain Weight with Whey Protein? Your Ultimate Guide
PFIC (Progressive Familial Intrahepatic Cholestasis)
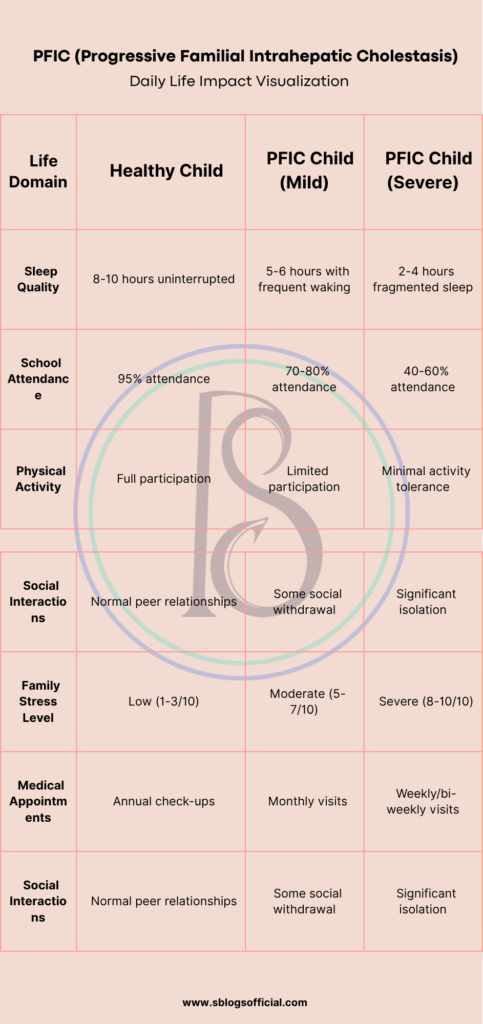
Progressive Familial Intrahepatic Cholestasis encompasses a group of rare inherited disorders that fundamentally disrupt the liver’s ability to transport bile acids and other substances from liver cells into bile ducts. These conditions result from genetic mutations affecting specific transport proteins essential for normal bile formation and flow, leading to a gradual accumulation of toxic substances within liver cells. The “progressive” nature of PFIC means that liver damage worsens over time, often beginning in infancy or early childhood and advancing relentlessly without appropriate intervention. Each type of PFIC involves different genetic defects and transport mechanisms, but all share the common pathway of impaired bile flow leading to cholestasis. Understanding PFIC is crucial because early recognition and treatment can significantly slow disease progression and improve quality of life for affected children.4
- What is it? A group of genetic disorders that affect bile flow from the liver, causing progressive liver disease in infants and children.
- Key symptoms: Severe itching (pruritus), jaundice, failure to thrive, fat-soluble vitamin deficiencies, enlarged liver and spleen.
- Why it matters: Without treatment, it progresses to cirrhosis and liver failure, often requiring liver transplantation in childhood.
- Clinical note: Three main types (PFIC1, PFIC2, PFIC3) with different genetic causes and severity levels, affecting approximately 1 in 50,000-100,000 births.
Alpha-1 Antitrypsin Deficiency
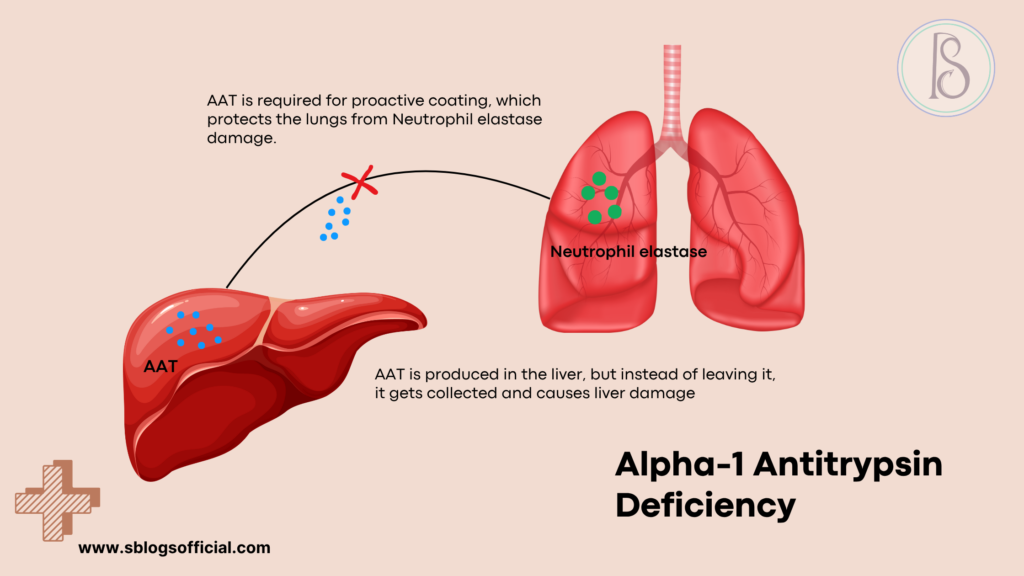
Alpha-1 antitrypsin deficiency stands as one of the most common genetic causes of liver disease in children, yet it remains significantly underdiagnosed due to its variable presentation and symptoms that can mimic other conditions. This inherited disorder results from mutations in the SERPINA1 gene, which normally produces alpha-1 antitrypsin—a crucial protein that protects tissues from damage caused by enzymes released during inflammation. When this protective protein is deficient or abnormal, it can accumulate in liver cells, causing progressive liver damage, while simultaneously leaving the lungs vulnerable to destructive enzymes. The condition demonstrates remarkable variability in its clinical presentation, with some individuals experiencing severe liver disease in infancy while others remain asymptomatic until adulthood. This genetic condition affects people of all ethnicities but shows particularly high prevalence in individuals of Northern European descent.5
- What is it? A genetic condition where the body doesn’t produce enough alpha-1 antitrypsin protein, leading to liver and lung damage.
- Key symptoms: In infants: jaundice, enlarged liver, slow growth; Later in life: breathing problems, liver cirrhosis, increased infection risk.
- Why it matters: It’s one of the most common genetic causes of liver disease in children and can lead to both liver and lung complications throughout life.
- Clinical note: Affects approximately 1 in 2,000-5,000 individuals, with over 100 genetic variants identified, most commonly the Z allele mutation.
Wilson Disease
Wilson disease represents a fascinating yet dangerous genetic disorder that disrupts the body’s natural copper balance, leading to toxic accumulation of this essential mineral in vital organs throughout the body. Named after British neurologist Samuel Wilson who first described the neurological aspects in 1912, this condition results from mutations in the ATP7B gene responsible for copper transport and elimination. Under normal circumstances, the liver processes dietary copper and eliminates excess amounts through bile, but in Wilson disease, this regulatory mechanism fails catastrophically. The accumulated copper acts like a slow poison, gradually damaging liver cells, brain tissue, and other organs over months to years. What makes Wilson disease particularly insidious is its ability to remain hidden for years before symptoms appear, often manifesting during childhood, adolescence, or early adulthood when copper levels reach critically toxic thresholds.6
- What is it? A rare genetic disorder that causes copper to accumulate in the liver, brain, and other organs due to defective copper transport.
- Key symptoms: In infants/children: liver enlargement, jaundice, fluid in abdomen; Later: neurological symptoms, psychiatric changes, Kayser-Fleischer rings in eyes.
- Why it matters: Without treatment, copper buildup causes progressive liver damage, neurological deterioration, and can be fatal.
- Clinical note: Affects approximately 1 in 30,000 people worldwide, caused by mutations in the ATP7B gene, with symptoms typically appearing between ages 5-35.
Neonatal Hemochromatosis
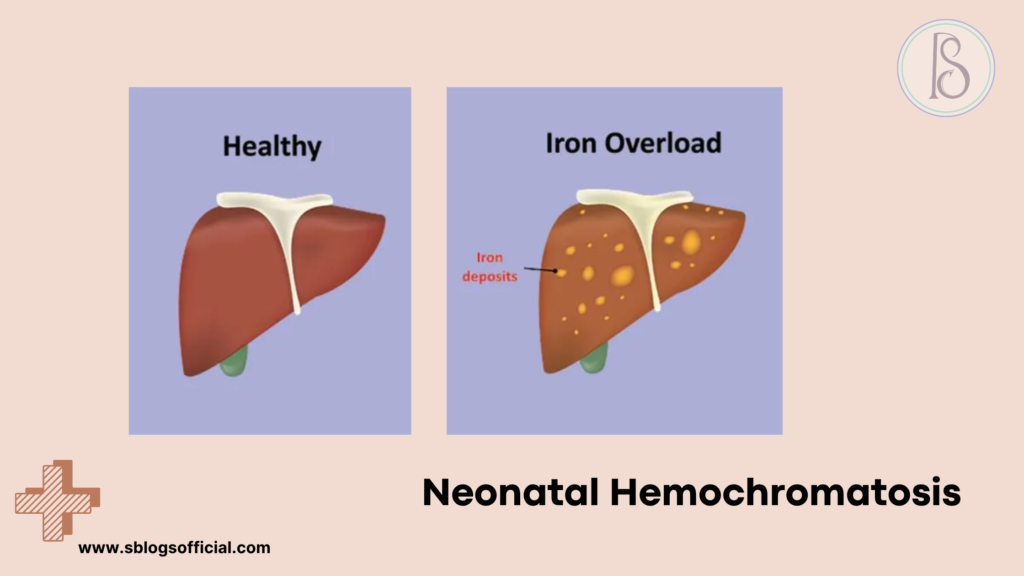
Neonatal hemochromatosis represents one of the most devastating and rapidly progressive liver conditions affecting newborns, characterized by severe iron overload that begins during fetal development. Unlike hereditary hemochromatosis seen in adults, neonatal hemochromatosis is now understood to be an immune-mediated condition where maternal antibodies attack the developing fetal liver, leading to catastrophic iron accumulation. This condition typically manifests within the first few days of life with fulminant liver failure, making it a true pediatric emergency requiring immediate recognition and intervention. The iron deposits primarily affect the liver but can also accumulate in the heart, pancreas, and other organs, creating a cascade of organ dysfunction. Understanding neonatal hemochromatosis is crucial because while historically fatal, recent advances in treatment protocols have dramatically improved survival rates when the condition is recognized and treated promptly.7
- What is it? A rare condition where iron accumulates in the liver and other organs of fetuses and newborns, causing severe liver damage.
- Key symptoms: Severe jaundice, low blood sugar, bleeding problems, heart failure, growth restriction, liver failure in first days of life.
- Why it matters: It’s often fatal without immediate treatment, but early recognition and treatment can be life-saving.
- Clinical note: Affects approximately 1 in 9,000 births, thought to be an immune-mediated process rather than a genetic iron overload disorder.
Galactosemia
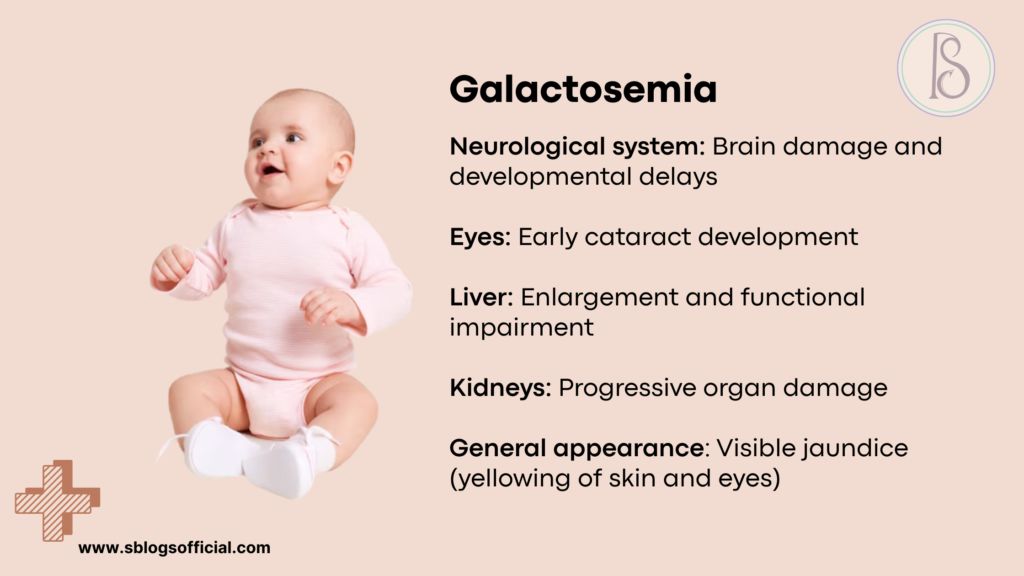
Galactosemia represents a critical genetic disorder that transforms milk—typically a source of essential nutrition for infants—into a potentially toxic substance that can cause severe organ damage. This inherited condition results from deficiencies in enzymes responsible for converting galactose, a sugar found in milk and dairy products, into glucose that the body can safely utilize. When these enzymes are absent or deficient, galactose and its metabolic byproducts accumulate to toxic levels, particularly affecting the liver, brain, kidneys, and eyes. The condition exists in several forms, with classic galactosemia being the most severe and requiring immediate dietary intervention to prevent life-threatening complications. Fortunately, galactosemia serves as one of the great success stories of newborn screening programs, as early detection and strict dietary management can prevent most of the devastating complications that once made this condition uniformly fatal or severely disabling.8
- What is it? A genetic disorder where the body cannot properly process galactose (a sugar found in milk), leading to toxic buildup affecting the liver and other organs.
- Key symptoms: Vomiting, diarrhea, failure to thrive, jaundice, enlarged liver, cataracts, developmental delays if untreated.
- Why it matters: Early detection and strict galactose-free diet can prevent severe complications including liver damage, intellectual disability, and ovarian dysfunction.
- Clinical note: Classic galactosemia affects 1 in 30,000-60,000 newborns, with newborn screening programs enabling early diagnosis and treatment.
Conclusion: Hope Through Awareness and Advances
The hereditary disorders we’ve explored in this first part—PFIC, Galactosemia, Alpha-1 Antitrypsin Deficiency, and Tyrosinemia—represent significant challenges for affected infants and their families. Each condition presents its own unique mechanisms of liver damage, symptomatic patterns, and treatment considerations. Early recognition remains the cornerstone of improved outcomes, making awareness of these conditions crucial for parents and healthcare providers alike.
While these disorders can be devastating, advances in genetic testing, specialised treatments, and liver transplantation techniques have dramatically improved prospects for many babies diagnosed with hereditary liver conditions. The journey from diagnosis to treatment may be complex, but families don’t have to navigate it alone.
In Part 2 of our series, we’ll continue exploring this important topic by examining Neonatal Hemochromatosis and other significant hereditary conditions affecting infant liver health. We’ll also provide comprehensive guidance on when to seek medical attention, current treatment approaches, and answer frequently asked questions about these challenging disorders. For the complete picture of hereditary disorders causing infant liver failure and crucial information that could help identify these conditions early, be sure to read the second installment of our series.
Source-
- Long-term outcome and necessity of liver transplantation in infants with biliary atresia are independent of cytokine milieu in native liver and serum – ScienceDirect ↩︎
- Tyrosinemia – an overview | ScienceDirect Topics ↩︎
- ↩︎
- Progressive Familial Intrahepatic Cholestasis – StatPearls – NCBI Bookshelf ↩︎
- Alpha-1 Antitrypsin Deficiency Epidemiology – Rare Disease Advisor ↩︎
- Wilson disease – Gastroenterology ↩︎
- Neonatal hemochromatosis: management, outcome, and prevention – Lopriore – 2013 – Prenatal Diagnosis – Wiley Online Library ↩︎
- Galactosemia: MedlinePlus Genetics ↩︎